


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
樋口 真理子; Pinak, M.
no journal, ,
放射線により損傷が10から20ベースペア以内に複数の損傷が生じることがあり、これをクラスター損傷と呼ぶ。クラスター損傷は単独損傷と比較して修復が阻害される傾向がある。8オキソグアニンの相補鎖上の数ベースペア離れた位置にAPサイトや一本鎖鎖切断がある損傷は修復が困難なクラスター損傷の例である。このクラスター損傷は修復酵素hOGG1による8オキソグアニンの修復を阻害することが実験によりわかっている。8オキソグアニンとの相対位置により修復率が変わるが、その原因はわかっていない。本研究では損傷DNAと修復酵素hOGG1の複合体の分子動力学シミュレーションを用いて修復阻害の仕組みの解明を目指した。APサイトあるいは鎖切断を複合体中のDNA上に配置し、場所を変えて数種類のシミュレーションを行った。その結果、鎖切断の位置によってDNAの構造揺らぎが変わることがわかった。損傷の位置によって酵素との接触面積が変わるからと考えられる。
山崎 智*; 河野 秀俊; 清水 謙多郎*; 皿井 明倫*; 寺田 透*
no journal, ,
Sequence-specific recognition of DNA by proteins plays a critical role in regulating gene expression. Accurate recognition is achieved by a combination of two different mechanisms. The first is direct readout, in which recognition is mediated by direct interactions between amino acids and DNA bases. The second is indirect readout, which is caused by the sequence-dependence of the conformation and deformability of the Drain the present study, we developed a new method for evaluating the contribution of indirect readout. This method uses the information entropy as a measure of the contribution. The entropy was calculated from the probability of DNA sequence given conformation, which was obtained by converting the probability of conformation given sequence using Bays'theorem. We used trajectories of MD simulations for free DNAs that had different tetramer sequences at the center of the sequences to calculate the probability distributions of the conformation. We found that the regions which are shown to be indirectly readout by experiment tend to have smaller information entropies. We therefore concluded that the information entropy is a good measure of the contribution of indirect readout in protein-DNA recognition.
新井 栄揮; 米澤 悌; 岡崎 伸生; 玉田 太郎; 徳永 廣子*; 石橋 松二郎*; 徳永 正雄*; 黒木 良太
no journal, ,
ヌクレオシドニリン酸キナーゼ(NDK)は、あらゆる生物に保存された蛋白質であり、オリゴマー構造を形成することが知られている。グラム陰性細菌由来のNDKは基本2量体が2つ集まった4量体、真核生物及び古細菌由来のNDKは基本2量体が3つ集まった6量体などの会合構造をとる。本研究では、中度好塩菌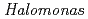 sp. 593株由来NDK(HaNDK)のE134A変異型の結晶構造を調べ、結晶構造中にグラム陰性菌
sp. 593株由来NDK(HaNDK)のE134A変異型の結晶構造を調べ、結晶構造中にグラム陰性菌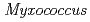 由来NDKに類似した4量体構造(Type I)と
由来NDKに類似した4量体構造(Type I)と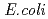 由来NDKに類似した4量体構造(Type II)が交互に現れることを明らかにした。この結果により、HaNDKが少ない変異導入によって多量体構造を容易に変化させることが示唆された。
由来NDKに類似した4量体構造(Type II)が交互に現れることを明らかにした。この結果により、HaNDKが少ない変異導入によって多量体構造を容易に変化させることが示唆された。
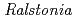 sp. A-471由来のGH Family 23に属する新規キチナーゼの構造解析
sp. A-471由来のGH Family 23に属する新規キチナーゼの構造解析玉田 太郎; 岡崎 伸生; 上田 光宏*; 中澤 昌美*; 宮武 和孝*; 黒木 良太
no journal, ,
好熱性細菌 sp. A-471由来の新規酵素(Ra-ChiC)の1.9 分解能での構造解析に成功した。Ra-ChiCの触媒ドメインは7本のへリックスから構成されており、うち5本へリックスについてはガチョウ型リゾチームの立体構造と類似していた。ガチョウ型リゾチームの触媒残基であるグルタミン酸残基はRa-ChiCにおいて側鎖を含め立体的によく保存されていた(Glu141)。ガチョウ型リゾチームで活性に関与していると考えられる2つのアスパラギン酸については、Ra-ChiCにおいて1つは立体的にほぼ似た箇所にグルタミン酸として存在していた(Glu162)のに対し、もう1つは主鎖構造から完全に異なっていた。これらの違いがキチナーゼ活性とリゾチーム活性の違いを創出していると考えられた。
分解能での構造解析に成功した。Ra-ChiCの触媒ドメインは7本のへリックスから構成されており、うち5本へリックスについてはガチョウ型リゾチームの立体構造と類似していた。ガチョウ型リゾチームの触媒残基であるグルタミン酸残基はRa-ChiCにおいて側鎖を含め立体的によく保存されていた(Glu141)。ガチョウ型リゾチームで活性に関与していると考えられる2つのアスパラギン酸については、Ra-ChiCにおいて1つは立体的にほぼ似た箇所にグルタミン酸として存在していた(Glu162)のに対し、もう1つは主鎖構造から完全に異なっていた。これらの違いがキチナーゼ活性とリゾチーム活性の違いを創出していると考えられた。
大原 高志; 安達 基泰; 清水 瑠美; 玉田 太郎; 黒木 良太; 西宮 佳志*; 近藤 英昌*; 津田 栄*
no journal, ,
不凍タンパク質(AFP)は生体内で氷の表面に結合することで氷結晶の成長を抑制し、体液の凝固点を下げる働きを持つタンパク質である。北海道沿岸に生息するナガガジの体内では多数の3型AFP(nfeAFP)のアイソフォームが発現しており、これらはSP型及びQAE型に分類される。どちらも氷表面に結合する機能は有するが、氷結晶の成長を抑制する能力はQAE型の方がはるかに高い。そこでわれわれは、SP型であるnfeAFP6とQAE型であるnfeAFP8に注目し、両者の間でアミノ酸配列が異なる部分についてnfeAFP6とnfeAFP8のキメラ体4種類と、nfeAFP6の配列をnfeAFP8のものに置換したnfeAFP6変異体8種類を調製して、これら変異体の存在下での氷単結晶の成長過程を顕微鏡観察することで氷結晶成長抑制作用を調べた。その結果、nfeAFP6のAla19一か所のみをValに置換したnfeAFP6 A19V変異体がQAE型と同様の氷結晶成長抑制作用を持つことを見いだした。さらにnfeAFP6 WT及びA19V変異体の両者について単結晶X線構造解析に成功した。本発表では、nfeAFP6 WTとnfeAFP6 A19Vの構造の違いについても併せて報告する。
栗原 和男; 玉田 太郎; 大原 高志; 黒木 良太
no journal, ,
水素原子及び水和水分子は、生体内反応において重要な役割を有す。特に水素原子は水素結合を通してタンパク質の立体構造形成や安定性に大きく寄与し、分子認識における重要な担い手でもある。われわれは生体反応や分子認識における水素や水和水の役割を解明するため、原子力機構研究用原子炉JRR-3に設置された2台の生体高分子用中性子単結晶回折装置(BIX-3, 4)を運用し、高度化を行っている。両装置では、年間に7 14構造の決定が可能であり、これまでに最高1.5分解能のデータ取得に成功している。また。例年、提供される175日間(2台で350日)のビームタイムのうち約30%に相当するビームタイムを施設共同利用に提供している。今回の発表では、2008年度から2009年度に実施した中性子回折実験(ブタ膵臓エラスターゼ,HIVプロテアーゼ他)の概要を紹介する。さらに近年では、同一の結晶から中性子回折データとX線回折データの両方を取得し、統合して1つの立体構造モデルを得るN/X同時精密化法を利用して構造決定を行っている。この手法の有効性について紹介する。2010年度からは、両装置に対し、新規の試料ゴニオメータと組合せた窒素吹付け型低温装置の導入を行う。100Kへの試料結晶冷却によるDebye-Waller因子の低減から、現在の常温測定より2倍以上の測定効率の向上を見込んでいる。
14構造の決定が可能であり、これまでに最高1.5分解能のデータ取得に成功している。また。例年、提供される175日間(2台で350日)のビームタイムのうち約30%に相当するビームタイムを施設共同利用に提供している。今回の発表では、2008年度から2009年度に実施した中性子回折実験(ブタ膵臓エラスターゼ,HIVプロテアーゼ他)の概要を紹介する。さらに近年では、同一の結晶から中性子回折データとX線回折データの両方を取得し、統合して1つの立体構造モデルを得るN/X同時精密化法を利用して構造決定を行っている。この手法の有効性について紹介する。2010年度からは、両装置に対し、新規の試料ゴニオメータと組合せた窒素吹付け型低温装置の導入を行う。100Kへの試料結晶冷却によるDebye-Waller因子の低減から、現在の常温測定より2倍以上の測定効率の向上を見込んでいる。
目黒 瑞枝; 安達 基泰; 岡崎 伸生; 玉田 太郎; 黒木 良太; 加藤 尚志
no journal, ,
エリスロポエチン(EPO)は、赤血球産生に必須な蛋白質である。われわれは、両生類で初めてツメガエルのEPOを同定することに成功した。遺伝子組換えxlEPOを調製し、ヒトEPO受容体を発現するUT-7/Epo細胞株を用いて増殖アッセイを実施したところ、生理活性は種を超えて交差することが判明した。この結果から、ツメガエルとヒトのEPOには、一次構造の類似性を超えて、特異的生物機能の発揮に必要となる特有の立体構造が共有されている可能性が考えられる。xlEPO組換え体を用いて、シッティングドロップ蒸気拡散法でxlEPO結晶を得ることに成功した。X線回折実験で2.9分解能の回折データを取得し、既に立体構造が報告されているヒトEPOの座標を用いた分子置換法によってxlEPOの立体構造を解析することに成功した。
安達 基泰; 新井 栄揮; 玉田 太郎; 黒木 良太; 日高 興士*; 木村 徹*; 木曽 良明*
no journal, ,
本研究では、ロピナビルに対して耐性を獲得したA17株ウイルス由来(A17型)HIV-1プロテアーゼの薬剤耐性獲得機構を原子レベルで解明し、より効果的な阻害剤の設計において有用な知見を得ることを目的として、ロピナビル及び耐性変異体に効果の高い阻害剤KNI-1657との複合体のX線結晶構造解析を実施した。A17型HIV-1プロテアーゼ遺伝子のコドン配列を大腸菌発現に最適化した人工遺伝子を合成し、野生型と同様な方法で調製した。得られた精製試料を用いて、A17型HIV-1プロテアーゼの活性発現に対するロピナビルの阻害効果を調べたところ、野生型に比較して700倍以上減少していた。一方で、KNI-1657はロピナビルよりも約20倍高い阻害効果を示した。A17型HIV-1プロテアーゼとロピナビル及びKNI-1657との複合体を結晶化した結果、PEG4000を沈殿剤とした条件で結晶化に成功した。得られた結晶を用いて、放射光施設(PF及びSPring-8)にて1.5分解能の回折データを収集し、R値19%まで構造を精密化した。
仲庭 哲津子*; 深田 はるみ*; 井上 達矢*; 木下 誉富*; 安達 基泰; 玉田 太郎; 黒木 良太; 多田 俊治*
no journal, ,
JNK1はストレス応答性のMAPキナーゼである。本研究はJNK1と阻害剤との複合体の構造をX線及び中性子回折法により超高分解能で決定し、構造情報に基づいた創薬法の新たな展開を図るものである。JNK1には構造内部に4か所と表面に3か所のCys残基があり、これらが酸化的条件下において失活や凝集を引き起こす原因となり、安定した巨大な結晶を作ることが困難であると考えられる。これまで本研究ではCysを変異したJNK1変異体を2種調製し、そのうち分子表面3か所のCysを変異させることで構造安定性を保持したまま大量発現が可能なM3を見いだした。しかし2種の変異体の構造解析より、両者ともに格子定数が100以上、分解能2.7程度と中性子回折に不適当であった。そこで既に報告されているJNK1アイソザイムの結晶系は中性子回折の条件を満たしていたことから、変異体との結晶パッキングの比較を行った。アイソザイムは変異型に比べて分子間の塩橋や水素結合が多いことがわかった。本発表ではアイソザイムのみで形成している相互作用2か所に注目し、それらをM3に導入した変異体のパッキング変化について報告する。
Fernandez, M.*; 藤井 聡*; 河野 秀俊; 皿井 明倫*
no journal, ,
Nucleosomes are the primary organizational units of chromatin in Eukaryotic chromosomes. They are composed of about 147 DNA base pairs wrapped around a histone octamer. Classical computational methods have limitations to analyze nucleosomes because of the weak sequence-dependent signal for nucleosome positioning. In general, proteins can recognize specific DNA sequences not only by direct contacts between amino acids and bases (direct readout) but also through conformational properties of DNA (indirect readout). Because of peculiar conformation of DNA in the nucleosome structure, indirect readout may play important role in nucleosome positioning along the genome. We calculated conformation energies of DNA by threading DNA sequences around crystal structures of nucleosomes. The strength of the conformation energy oscillates at frequency 10 bps for dinucleotides, in agreement with the previous nucleosome models, in which distinctive sequence motifs recurring periodically at the DNA helical repeat are known to facilitate the sharp bending of DNA around the nucleosome. The DNA conformation energy calculated along yeast genome positively correlated with nucleosome positioning experimentally measured.